
NUTRIENT
ANALYSIS METHODS MANUAL
FOR HUMIC-CONTAINING ESTUARINE WATERS
PHOSPHATE
Initially we tried the same method but with the usual antimony in reagent A (standard method based on Murphy and Riley (1962) method)(D'Elia et al., unpubl.). Carryover between samples (tailing of peaks) was extensive. Samples tended to both "wash in" and "wash out" slowly, so that there was loss of some samples and enhancement of others depending on surrounding samples. (The colloidal reaction product tends to stick to tubing walls or spectrophotometer cuvettes.) Sensitivity was greater but accuracy and precision were much worse. We tried incorporating some ethanol into reagents to counteract sticking (Edwards et al., 1965), but that did not improve tailing significantly. Loss of sensitivity without antimony could be overcome by increasing Standard Calibration setting. We also experimented with various color blank reagents, as some authors suggest leaving out ammonium molybdate (Koroleff, 1983) or ascorbic acid (APHA Standard methods..., 1981) or both (Flebbe, 1982; Parsons, et al., 1984).
SAMPLES
- Sample Volume:
- 12 mL
- Sample Collection:
- Filter through ashed GF/F filter in glass or plastic apparatus,
ashed or used only for similar samples.
- Sample Storage:
- in polyethylene bottle at -20 oC or lower
REAGENTS
- 4.9 N H2SO4:
- Carefully (slowly and with stirring) add 136 mL concentrated
(sp. gr. = 1.84 g cm-³) H2SO4
to approx. 800 mL water. Cool and dilute to 1 L with water.
- Ammonium Molybdate:
- Dissolve 40 g (NH4)6Mo7O24.4H2O
and make up to 1 L with water. Store in plastic bottle in dark.
- Ascorbic Acid:
- Dissolve 18 g C6H8O6 and
make up to 1 L with water. Dispense 40 mL aliquots into plastic
bottles and freeze.
- SLS:
- Dissolve 3 g sodium lauryl sulfate (sodium dodecyl sulfate,
phosphate < 0.0001%) and make up to 100 mL with water.
- Working Reagent A:
- Combine (in this order) 50 mL 4.9 N H2SO4,
15 mL ammonium molybdate, and 1 mL SLS. Make fresh every few
days.
- Color Blank Reagent A:
- Combine 50 mL 4.9 N H2SO4, 15 mL water,
and 1 mL SLS.
- Working Reagent B:
- Thaw 40 mL ascorbic acid solution and add 0.4
mL SLS. Make fresh daily.
STANDARDS
- 5 mM Primary:
- Dry KH2PO4 overnight at 110 oC.
Dissolve 0.68 g and make up to 1 L with water. Preserve with a few drops of
chloroform and store in refrigerator.
- 250 uM Secondary:
- 0.4 mL Primary Standard + 7.6 mL water, or similar convenient
amount. Make fresh each day.
- Working:
- Make fresh each day:
- 1 uM: 3.984 mL water + 16 uL Secondary Std.
- 2 uM: 3.968 mL water + 32 uL Secondary Std.
- 5 uM: 3.920 mL water + 80 uL Secondary Std.
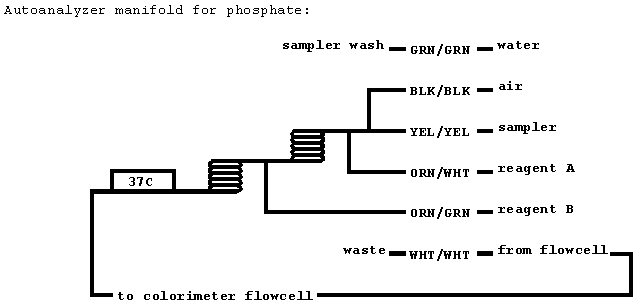
SPECIAL EQUIPMENT
- Technicon AutoAnalyzer II with
phosphate manifold
(reproduced from D'Elia et al., unpubl.), 50 mm x 1.5 mm
flowcell, 880 nm filters and phototubes 199-B021-04. Heating
the reaction coil to 70 oC if possible may be desirable
(Hansen and Grasshoff, 1983). 820-830 nm filters may be desirable if
available.
{kind=link}
PROTOCOL
Following D'Elia et al. (unpubl.), based on Technicon Industrial Method no. 155-71W but with the single reagent split into two for better stability. One important difference from D'Elia et al. is that antimony is omitted to reduce tailing of sample peaks (Hansen and Grasshoff, 1983). A color blank procedure is used to correct for humic color (Koroleff, 1983). Dissolved inorganic phosphate is determined.
- Autoanalyzer Set-Up:
Allow colorimeter and chart recorder to warm up for approx. half an hour. Set zero and full scale on chart recorder relative to colorimeter. Set Standard Calibration setting 9.5, Damp setting "normal", Sample Rate 40/hr, Sample:Wash 9:1, Chart Speed 30 cm/hr. Pump with water in sample line (i.e. sampler off) and reagents in reagent lines until baseline stabilizes. Set baseline to desired reading on chart recorder using the baseline knob on the colorimeter. (Set to some low positive value, such as 5% full scale, rather than zero to allow for any negative drift.)
Make up triplicate working standards in test tubes, 4 mL each. Fill (use approx. 4 mL) polyethylene autoanalyzer cups set up in sampler tray in the following order (all in triplicate except where noted): 5 uM std., 2 uM std., 1 uM std., water, 2 samples, 1 cup water, 2 samples, 1 cup water, 2 samples, 1 cup water, 2 samples. Fill second tray likewise putting 1 cup water between every 2 triplicate samples.
Run standards and samples for phosphate + color.
Run standards and samples for color blank: Replace reagent A with color blank reagent A and allow baseline to stabilize. Do NOT reset baseline. Standards may require a very slight color correction. Note that color blanks do not correlate with visible color of samples. "Color" at this wavelength may be due to materials other than humics, so always run color blanks.
- Autoanalyzer Shut-Down:
- Pump with water in sample and reagent
lines for approx. 10 min. Shut off all components and
disengage pump.
CALCULATIONS
Baseline shifting may be severe. Use water cups between samples, as well as beginning and ending baseline levels, to interpolate baseline for each individual peak for both phosphate + color and color blank runs.
Read the top of each peak. There are often sudden "spikes" at the tops of peaks. They seem to be due to effects of seawater and freshwater mixing at the interface between sample and wash: ignore them and read the flat tops of the peaks.
"Adjusted peak height" = top of peak - baseline
"Corrected peak height" = "Adjusted peak height for PO4 + color" - "Adjusted peak height for color blank"
Plot "Corrected peak height" of standards vs. concentration. Obtain the slope and intercept of this line.
Sample concentration (uM) = (Sample "Corrected peak height" - intercept) ÷ slope
No correction for salinity is necessary.
Silicate interference is equivalent to approximately 0.05% of its concentration (i.e. in two trials, 100 uM silica appeared as 0.03 and 0.08 uM phosphate) due to a slower reaction speed and non-optimum reaction pH. Silicate concentrations in Georgia rivers may be high; however, reactive silicate concentrations in samples that have been frozen and thawed just prior to use are much lower than concentrations in unfrozen samples, probably due to polymerization during freezing. Interference as apparent phosphate may be on the order of 10% or less of the actual phosphate. Therefore, silicate interference may be ignored in samples that are frozen and thawed just prior to phosphate analysis.
Arsenate interference is equivalent to approximately 10% of its concentration (i.e. 1 uM arsenate appears as approximately 0.1 uM phosphate) due to a slower reaction speed. Therefore it can be ignored unless arsenate is suspected to be very high.
- Limit of Detection:
- Due to often extreme baseline shifting, blanks in
this analysis are defined as zero; therefore, their std. dev. =
0. To estimate limit of detection, use std. dev. of the lowest
standard. 2 std. dev. above blank approx. = 0.03 uM
- Range:
- approximately 100 uM by changing Standard Calibration setting.
- Accuracy:
- 95% confidence limits for prediction of a concentration
near the mean for a typical standard curve (2 uM) approx. =
± 0.38 uM for a triplicate determination. (Confidence limits for
prediction near the ends of a standard curve are, of course,
somewhat larger.)
REFERENCES
- American Public Health Association Standard methods for the
examination of
- water and wastewater: 15th edition. 1981.
American Public Health Association, Washington, D.C.
- D'Elia, C.F., N.L. Kaumeyer, C.L. Keefe, D.L. Shaw, K.V. Wood,
and C.F.
- Zimmermann. 1987 unpubl. Standard operating
procedures. Nutrient Analytical Services Laboratory,
Chesapeake Biological Laboratory, Solomons, Maryland.
- Edwards, G.P., A.H. Molof, and R.W. Schneeman. 1965.
Determination of
- orthophosphate in fresh and saline waters.
J. Amer. Wat. Wks. Ass. 57: 917-925.
- Flebbe, P. 1982. Biogeochemistry of carbon, nitrogen, and
phosphorus in the
- aquatic subsystem of selected Okefenokee
Swamp sites. Okefenokee Ecosystem Investigations no. 8.
- Hansen, H.P., and K. Grasshoff. 1983. Procedures for the
automated
- determination of seawater constituents. Pages
362-379 in Grasshoff, K., M. Ehrhardt, and K. Kremling,
editors. Methods of seawater analysis: second, revised and
extended edition. Verlag Chemie, Weinheim.
- Koroleff, F. 1983. Determination of phosphorus. Pages 125-131
in Grasshoff, K.,
- M. Ehrhardt, and K. Kremling, editors.
Methods of seawater analysis: second, revised and extended
edition. Verlag Chemie, Weinheim.
- Murphy, J., and J.P. Riley. 1962. A modified single solution
method for the
- determination of phosphate in natural waters.
Anal. Chim. Acta 27: 31-36.
- Parsons, T.R., Y. Maita, and C.M. Lalli. 1984. A manual of
chemical and biological
- methods for seawater analysis.
Pergamon Press, New York.
![]()
![]()
![]()
![]()
![]()